
In 1993 it was discovered that mutations in the gene encoding the enzyme superoxide dismutase 1 (SOD1) can cause amyotrophic lateral sclerosis (ALS), a neurodegenerative disease paralyzing the muscles of the body. The same year, a research collaboration focusing on ALS was established at Umeå University and University Hospital of Northern Sweden (NUS). The participating researchers were Stefan Marklund at the Department of Clinical Chemistry, Thomas Brännström at the Department of Clinical Pathology, and Peter Andersen at the Department of Clinical Neurology, and the aim was to study SOD1 in relation to ALS to understand how the disease is caused.
Since then, the research collaboration has grown and evolved into a network of research groups at Umeå University, sharing expertise and infrastructure to make new discoveries about ALS. Our aim is to contribute with knowledge to facilitate for future treatments of the disease.

ImageIllustration created by Ulrika Nordström using BioRender.com.
We have now built the largest biobank in Sweden with samples for ALS research and is the leading Swedish center for several clinical trials. We are running research projects in biochemistry, genetics, histopathology, cell biology, anatomy and clinical neurology, including pharmaceutical studies with ALS patients.
Together we have published over 200 ALS-related studies, and the collaboration spans around 25 coworkers dedicated to ALS research, among them laboratory assistants, PhD-students, post-docs, research engineers, research nurses, physicians, and professors. The researchers in the group have backgrounds in several different specialties, such as neurology, chemistry, pathology, molecular biology, and genetics.

Research nurse Lena Bylund, doctoral student in neurophysiology Ivar Winroth, research nurse Nina Sundqvist, senior neurologist and professor Peter Andersen, research nurse Elisabeth Müller Granberg, research nurse Tina Björn, and neurologist and associate professor Karin Forsberg make up the ALS team at the research floor of the neurology clinic at University Hospital of Northern Sweden.
Image Mattias Pettersson
Research nurse and study coordinator Lena Bylund handles research samples arriving for analysis.
Image Mattias Pettersson
Research nurse and study coordinator Tina Björn by her computer at the clinic.
Image Mattias Pettersson
Research nurse and study coodinator Nina Sundqvist is discussing something with doctoral student and physicist Ivar Winroth in her office.
Image Mattias Pettersson
Research nurse and study coodinator Elisabeth Müller Granberg in the process of taking a blood sample.
Image Mattias Pettersson
Blood samples are taken both for use in the ALS team’s own research, and as part of clinical trials held in collaboration with pharmaceutical companies.
Image Mattias Pettersson
Research nurse and study coodinator Nina Sundqvist is using a spirometer to measure pulmonary function.
Image Mattias Pettersson
Ivar Winroth, resident in neurophysiology and doctoral student at the Department of Clinical Sciences, is performing the standardised cognitive test ECAS, that many patients are subjected to while being assessed for ALS.
Image Mattias PetterssonBeing active at the university hospital of the Northern healthcare region and the largest hospital in the region of Västerbotten (Region Västerbotten), we receive many patients with suspected ALS for diagnostic testing and evaluation. We supply genetic counceling to neurological clinics and patients, as well as perform genetic testing for SOD1-mutations in ALS patients from all over the world. We are also the leading Swedish study center for several pharmaceutical studies.
The following clinical trials are ongoing and currently recruiting new patients:
The following trials are ongoing at University Hospital of Northern Sweden (NUS) but no longer recruting new patients:
Several new trials are under way, either in the planning stage or under negotiation with the pharmaceutical industry, the Swedish Medical Products Agency, and the Swedish Ethical Review Authority to be permitted to be held in Sweden. We will post updated information if, and when, the studies have been approved by the Swedish Ethical Review Authority.
Suggested person of contact: Peter Andersen and Lena Bylund
Affiliated personel: Peter Andersen, Karin Forsberg, Lena Bylund, Tina Björn, Elisabeth Müller Granberg, Nina Sundqvist and Ivar Winroth
Through the years, we have gathered more than 14,000 samples from ALS patients and their families. With the help of these samples we, in different collaborations with others, have discovered changes in several genes that can increase or decrease the risk of developing ALS. Our focus for many years has been ALS caused changes in the SOD1 gene. By analyzing our collected samples, we have found around 20 new mutations in the SOD1 gene in ALS patients, among them the D90A-mutation that is the most common SOD1 mutation in the Scandinavian population.

Sequencing data from a sample from a patient or research person. The analysis can show whether there is a mutation known for causing ALS.
ImageMattias PetterssonIn addition, we have helped develop a clinically useful method for identifying mutations in the C9orf72-gene, the most common genetic change known today that can cause ALS. We routinely screen for mutations in the C9orf72 and SOD1 genes in all ALS patients that are being treated by us, that have blood samples sent for analysis by their own treating physician, or that wish to participate in the European collaboration Project MinE, where the entire genome of over 350 of our test subjects have been sequenced for so called whole genome analysis. The aim is to study and compare genetic changes in thousands of ALS patients across Europe, Australia, and the USA to identify new leads to what can cause or protect against ALS.
Suggested persons of contact: Peter Andersen, Angelica Nordin
Affiliated personel: Peter Andersen, Angelica Nordin, Helena Alstermark, Eva Jonsson, Ivar Winroth
Our biochemical studies span cellular metabolism, protein function, and signaling between cells. The primary focus is on how different proteins involved in disease development are regulated, assembled and degraded, assume their three-dimensional form, and function.

Illustration showing the normal structure of the SOD1 protein.
ImageShutterstock, Copyright (c) 2018 Shutterstock. No use without permission.Since we first started out in 1993, we have extensively studied the SOD1 protein and how it causes ALS. Through the years we have learned that the enzymatic, normal function of SOD1 is sometimes altered in patients with mutations in the gene. However, this does not seem to be the primary cause of SOD1-related ALS, since mutations not altering the protein’s ability to clean up harmful superoxides can also cause ALS. Instead, the mutations seem to negatively affect the stability of the protein. The result is a protein that can “lose” its three-dimensional structure, and assume new, faulty structures. These new forms of so called “misfolded SOD1” aquire disease-causing qualities and can, in addition, spread between different cells and force other SOD1 proteins to assume the same pathogenic form (disease associated structure).

Illustration of how misfolded SOD1 proteins can look when they start forming the protein fibrills that accumulate and form the characteristic SOD1 lumps seen in nerve cells damaged by ALS. Some parts of the protein are locked away in the core of the fibrill. Others protrude and can be bound by e.g., therapeutic antibodies.
ImageShutterstock, Copyright (c) 2017 Shutterstock. No use without permission.In our biochemistry laboratory, we measure enzyme activity, analyse misfolded SOD1, and try to understand the underlying chemical processes transforming the protein from its correct, functional form to the misfolded, pathogenic form.
Suggested persons of contact: Per Zetterström, Stefan Marklund
Affiliated personel: Helena Alstermark, Eva Jonsson, Agneta Öberg, Karin Hjertkvist, Ulrika Nordström, Caitin Henne, Laura Leykam
In short, biomarkers are measurable substances, such as proteins, lipids, or other molecules, that vary in levels or leak out into the bloodstream or spinal fluid during disease. We collect and examine blood and spinal fluid samples from patients to see how different molecules and proteins are affected by the disease.

Research samples from patients and family members are aliquoted and can be used for many different types of analyses. There are generally large individual differences, which means we have to collect samples from many patients with the same type of ALS to make safe conclusions. Primarily, blood plasma, red and white blood cells, and cerebrospinal fluid are analyzed.
ImageMattias PetterssonThe aim is to find molecular changes that can act as clinically meaningful markers that can facilitate diagnosing ALS, predict disease progress, and indicate whether a drug is effective in slowing down the disease.
Suggested persons of contact: Peter Andersen, Arvin Behzadi
Affiliated personel: Helena Alstermark, Eva Jonsson, Agneta Öberg, Karin Hjertkvist
In pathology studies, cells, tissues, and organs, are analyzed in order to diagnose and learn more about diseases. We can not take samples to study changes in motor neurons in living patients without damaging the nervous tissue. To study the afflicted cells in the spinal cord and brain, ALS research is dependent on patients, and their next of kin, being in contact with us while alive and having expressed a wish to donate their bodies to research after death.
Through research autopsies, we can verify after the fact that ALS was correctly diagnosed. We can also learn a lot about the impact of the disease on the nervous system by studying the brain, the spinal cord, and the muscles of deceased patients. An important discovery made is that there are accumulations of misfolded or mislocated proteins in the afflicted cells in spinal cord tissue.

Paraffin-embedded tissue is sectioned and mounted on a glass slide for subsequent immunohistochemistry staining and analysis.
ImageMattias PetterssonThis knowledge has, among other things, lead to the development of treatment strategies targeting these harmful accumulations of misfolded proteins.
One of our most notable discoveries is that most ALS patients, whether they carry a mutated SOD1 gene or not, have aggregates of misfolded SOD1 in their nerve cells. This indicates that SOD1 influences the disease process in many more patients than simply those who carry specific SOD1 mutations.
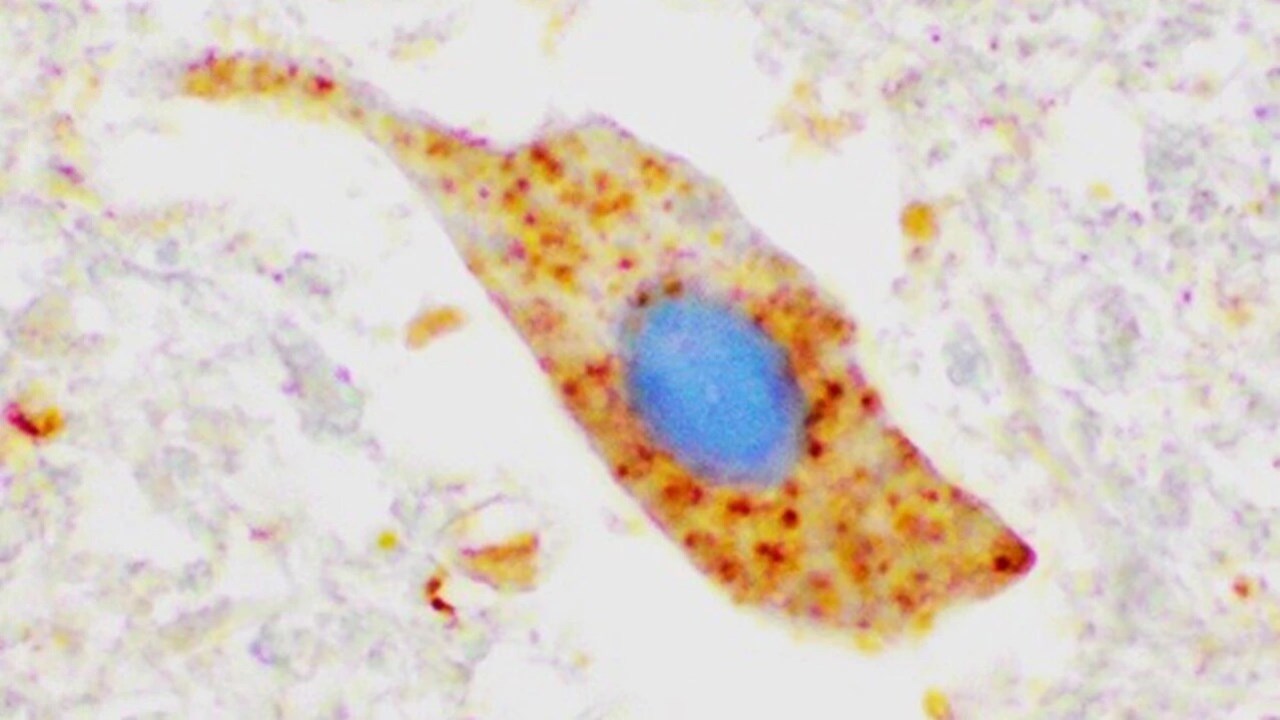
The image shows lumps of SOD1 protein (dyed brown) in a motor neuron in spinal cord tissue from an ALS patient not carrying a SOD1 mutation. This indicates that the protein may be involved in disease development even in sporatic ALS.
ImageKarin ForsbergSuggested persons of contact: Thomas Brännström och Karin Forsberg
Affiliated personel: Matthew Marklund, Sara Rimpi, Isil Keskin, Erica Stenvall, Isabelle Sigfridsson, Kornelia Åman Grönlund
An incomprehensible feature of ALS is that certain muscles in the body are much less affected by the disease. Eye muscles are an exemple of that. Even if some patients, after long respiratory care, may develop symptoms of eye paralysis, most patients – whether the disease started in the face or in the extremities – are relatively unaffected in their eye movements.

The motor nerve cells controlling the movement of the eyes are especially resilient in ALS. By studying these cells, we hope to find clues to specific properties that can prevent or slow down disease development.
ImageRuben Joye/MostphotosThe eye muscles are highly specialized, evolutionary conserved muscles with properties that no other muscles have. If the preservation of eye muscle function in ALS can be better understood, it may be possible to find new ways of treating the disease. Our studies have shown that the eye muscles in deceased ALS patients are relatively well preserved compared to other muscles, but that there are subtle signs of disease. The disease seems to give rise to changes in the signaling between eye muscles and their nerve cells, and different types of eye muscle fibers seem to be differently affected by the disease, but so far it is not possible to say which property, or properties, that makes them so resilient in ALS.
Suggested persons of contact: Fatima Pedrosa Domellöf, Jingxia Liu
Since we can not study changes in the motor nerve cells in the brain and spinal cord of living patients without harming the nervous tissue, we need models for studying disease-associated processes that can cause ALS. Through the years, we have collected skin biopsies and cultivated fibroblasts, i.e., connective tissue cells or supporting cells from the skin, from ALS patients ande their family members. These cells can be kept frozen to later be thawed and grown as cell cultures in laboratories.

Cells from a skin biopsy can be used to study the disease process in ALS outside the patient's own body.
ImageMattias PetterssonThrough cell culture experiments we can study how the disease affects cells from ALS patients and how the cells and disease-associated proteins react under different conditions. We have been able to study how the genetics of the patient and the environment of the cell interact and influence disease-associated factors, e.g., the accumulation of misfolded SOD1. We have also transformed some of our cell lines to stem cells and made those mature into motor nerve cells. This allows us to study the disease process in cells outside the patient’s own body. Cell culture experiments have given us insight into which factors contribute to increased misfolding, but also into which systems inside the cell that are important for breaking down misfolded SOD1.
Suggested persons of contact: Peter Andersen, Ulrika Nordström, Jonathan Gilthorpe
Affiliated personel: Eva Jonsson, Isil Keskin, Chloe Williams
To better understand the disease development in ALS, we conduct animal experiments within the framework of Umeå Centre for Comparative Biology (UCCB). We have produced several genetically altered mouse strains that express human SOD1 with different mutations capable of causing ALS in humans. Expression of mutated SOD1 protein results in an ALS-mimicking disease in these mice, much similar to the one afflicting humans.
The access to several different mouse strains allows us to study different stages of the disease progress in the nervous system in a way that would be impossible in patients. The differences between the mouse strains also reflects the corresponding differences between different patient groups, which helps us investigate why the disease progression is sometimes more aggressive and sometimes slower.
Through injection experiments we have also created a mouse model for studying the spread of aggregated misfolded SOD1. The model allows us to study the effect and spread of protein aggregates and their role in disease development.
All research involving laboratory animals has been approved by The Regional Ethics Committee and is performed in accordance with Swedish laws and EU directives and regulations concerning the use of animals for scientific purposes.



